

From L to R: Dr. Arko Barman, Jake Patock, and Dr. Rinki Ratnapriya.
A collaborative study by researchers from Rice University and Baylor College of Medicine has been recognized with the Best Paper Award at the Institute of Electrical and Electronics Engineers (IEEE) International Conference on Data Mining (ICDM), one of the field’s premier conferences. The study—conducted by Jake Patock, a former master’s student in data science at Rice, under the guidance of Arko Barman, associate teaching professor at Rice’s Data to Knowledge (D2K) Lab, and Rinki Ratnapriya, assistant professor of ophthalmology at Baylor—was presented at the workshop on Foundation Models for Biology and Bioinnovation in Washington, D.C., this November.
The study introduces a new method to reliably identify functional gene clusters associated with age-related macular degeneration (AMD), a genetically inherited degenerative eye disease that causes loss of sharp vision and can lead to complete blindness.
Traditionally, researchers study individual gene expression profiles to understand how they function. However, genes rarely act alone; they function in concert with other genes. Understanding how changes within these functional gene clusters contribute to disease is key to developing new treatments to prevent symptom onset or halt its progression.
To address this challenge, the researchers developed a three-step, graph-based method to create three-dimensional representations of these interactions for accurate and easy data analysis and visualization of 81 genes known to be responsible for AMD.
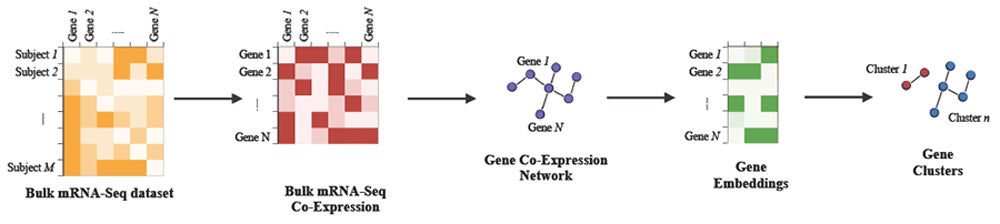
The first step involved creating a visual map of the co-expression network where each gene was represented as a node. Genes that are active or "expressed" together were linked by weighted lines indicating the strength of their relationship. Next, an embedding algorithm was used to transform each gene’s activity into a numerical representation. Finally, these numerical representations were then grouped to distinct gene clusters that act together in a specific pathway.
“An important feature that differentiates this method is that all steps are jointly fine-tuned using a special type of optimization algorithm, which ensures the results are reliable and robust every time,” Barman said. “Our method can be applied broadly to map functional gene interactors and networks for many genetic disorders, which can then serve as genetic signatures (aka biomarkers) for diagnosis and therapies.”